海洋所揭示蒽环类药物甲基转移酶功能调控“分子开关”
近日,中国科学院海洋研究所海洋生物制造研究组在蒽环类抗肿瘤药物的生物合成机制研究方面取得关键突破。团队成功揭示了两种同源甲基转移酶(DnrK与RdmB)的功能分化机制,发现了控制其催化功能在“脱羧”与“羟化”之间精准切换的关键分子开关,并由此获得了抗癌活性显著优于经典药物阿霉素的新型蒽环类衍生物。相关研究成果发表在催化领域国际期刊ACS Catalysis。
蒽环类药物(如阿霉素、表阿霉素)是临床肿瘤化疗的基石,但其严重的心脏毒性限制了其广泛应用。长期以来,科学界希望通过结构改造优化其疗效、降低毒副作用。这类结构复杂的分子在自然界中由一系列生物酶逐步催化合成。值得关注的是,参与蒽环抗生素合成的甲基转移酶在进化中出现了功能分化,其中DnrK(参与阿霉素合成)与RdmB(参与β-紫红霉素合成)尤为特殊:两者三维结构高度相似,序列同源性达53.4%,却能催化底物在C10位发生截然不同的反应——DnrK主要催化10-脱羧,而RdmB主要催化10-羟化。这种“同源不同功”的现象是蒽环类抗生素结构多样性的重要来源,但其背后的分子机制一直未被阐明。
研究团队综合运用结构生物学、生物化学及酶理性工程等手段,最终锁定DnrK蛋白α-16螺旋上的一个谷氨酸残基(E299)为调控催化路径的关键“开关”。只需对该位点进行精准替换,即可实现酶功能的定向翻转:将DnrK中的E299替换为疏水性氨基酸(如丙氨酸A或亮氨酸L),酶便获得类似RdmB的羟化功能,生成产物4;反之,将RdmB中对应位置的疏水氨基酸亮氨酸(L)替换为谷氨酸(E),则可使其获得类似DnrK的脱羧功能,生成产物2。
基于这一发现,研究人员进一步构建出能够同时催化脱羧与羟化反应的“杂合酶”,实现了两类蒽环衍生物的同步合成。活性测试表明,所获得的化合物2和4对人慢性髓性白血病K-562细胞具有强效抑制活性,其抗癌效力分别达到临床常用药阿霉素的15倍和20倍。
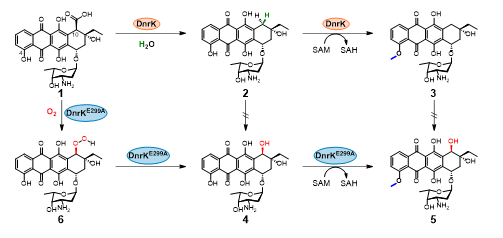
图1 DnrK及其突变体催化反应路径
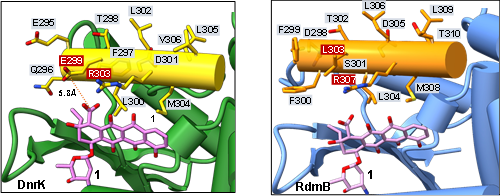
图2 DnrK与RdmB催化口袋关键氨基酸比对,其中关键调控位点氨基酸由红色高亮标注
该研究阐明了同源甲基转移酶功能分化的结构基础,揭示单个氨基酸残基对酶催化行为的决定性调控作用,彰显了酶理性工程在药物骨架定向修饰与优化中的巨大潜力。该研究不仅为理解自然界复杂药物分子的进化过程提供了关键见解,也为利用合成生物学技术创制新一代活性更高、毒性更低的蒽环药物奠定了重要的理论与技术基础。
中国科学院海洋研究所张伟研究员为论文通讯作者,博士后桑茉莉为第一作者。研究得到了国家自然科学基金、中国科学院海洋研究所自主部署项目、中国博士后科学基金等项目的支持。
论文信息:
Moli Sang, Qingyu Yang, Jiawei Guo, Peiyuan Feng, Yu Gao, Wencheng Ma, Shengying Li, Mikko Metsä-Ketelä, Wei Zhang*, Functional Plasticity of Methyltransferases in Anthracycline Biosynthesis: A Single Residue Reversal between Decarboxylation and Hydroxylation. ACS Catal. 2026. https://doi.org/10.1021/acscatal.5c07819
附件下载:

 鲁公网安备37020202001323号
鲁公网安备37020202001323号 | 古镇口园区地址:青岛市西海岸新区海军路88号 南海路园区地址:青岛市市南区南海路7号 科考船码头基地:青岛市西海岸新区长江东路8号 |
邮编:266000 邮件:iocas@qdio.ac.cn 电话:0532-82898611 传真:0532-82898612 |


